
Artificial intelligence method promptly predicts how two proteins will connect | MIT Information
4 min read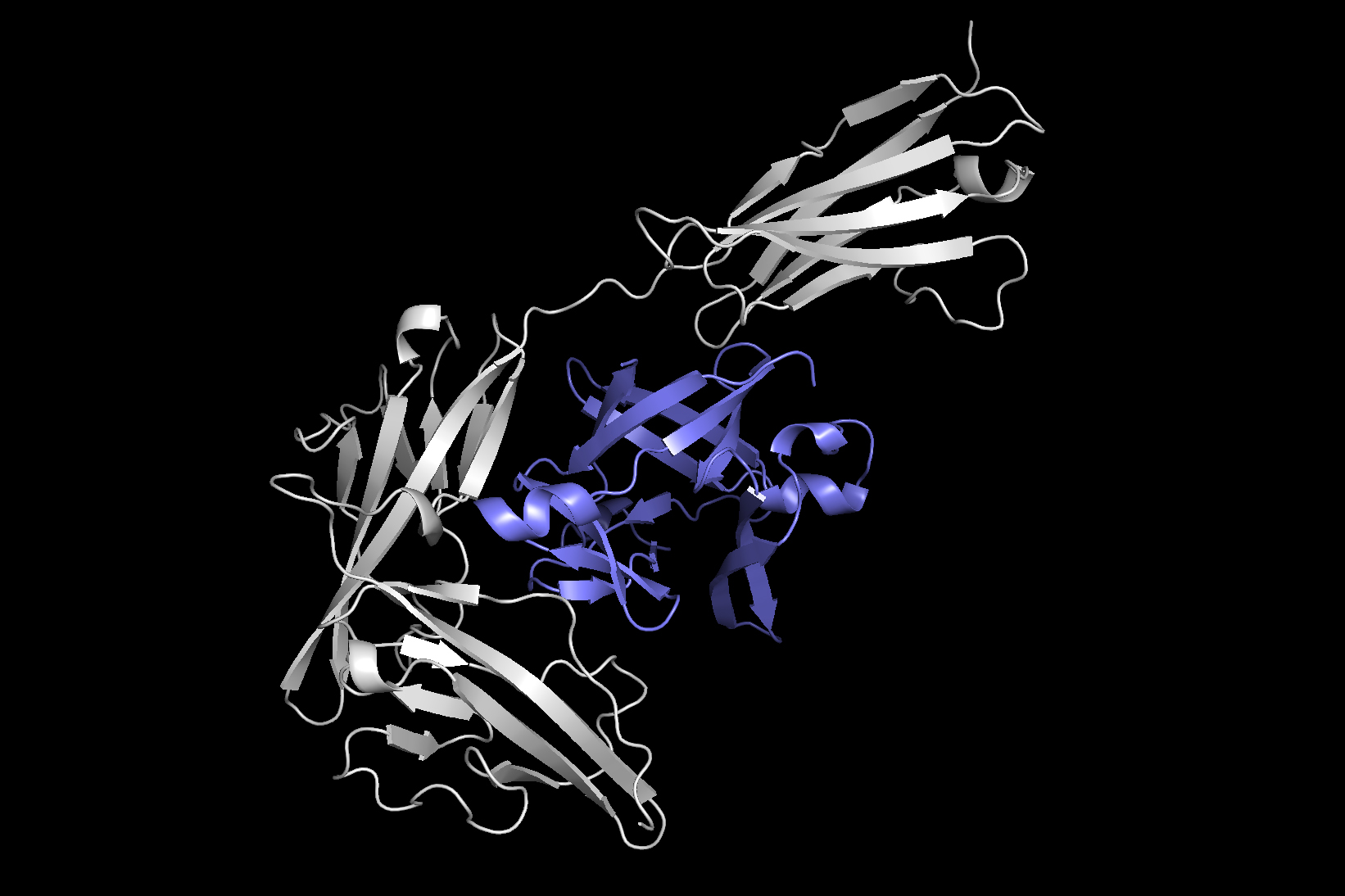
Antibodies, tiny proteins developed by the immune procedure, can attach to precise components of a virus to neutralize it. As scientists go on to battle SARS-CoV-2, the virus that results in Covid-19, a single feasible weapon is a artificial antibody that binds with the virus’ spike proteins to protect against the virus from moving into a human cell.
To build a profitable artificial antibody, scientists ought to fully grasp specifically how that attachment will happen. Proteins, with lumpy 3D buildings made up of a lot of folds, can adhere jointly in hundreds of thousands of combos, so locating the right protein intricate amid almost countless candidates is very time-consuming.
To streamline the approach, MIT scientists created a device-mastering design that can directly predict the complex that will form when two proteins bind jointly. Their technique is amongst 80 and 500 times speedier than point out-of-the-art program solutions, and frequently predicts protein structures that are nearer to precise constructions that have been observed experimentally.
This procedure could enable scientists far better comprehend some organic procedures that require protein interactions, like DNA replication and mend it could also speed up the method of building new medications.
“Deep learning is incredibly good at capturing interactions among distinctive proteins that are or else hard for chemists or biologists to produce experimentally. Some of these interactions are extremely sophisticated, and folks haven’t discovered great techniques to categorical them. This deep-studying model can find out these types of interactions from knowledge,” says Octavian-Eugen Ganea, a postdoc in the MIT Personal computer Science and Artificial Intelligence Laboratory (CSAIL) and co-lead creator of the paper.
Ganea’s co-guide creator is Xinyuan Huang, a graduate pupil at ETH Zurich. MIT co-authors consist of Regina Barzilay, the Faculty of Engineering Distinguished Professor for AI and Wellbeing in CSAIL, and Tommi Jaakkola, the Thomas Siebel Professor of Electrical Engineering in CSAIL and a member of the Institute for Facts, Methods, and Culture. The investigate will be offered at the Worldwide Convention on Learning Representations.
Protein attachment
The design the scientists developed, called Equidock, focuses on rigid entire body docking — which happens when two proteins connect by rotating or translating in 3D place, but their styles really don’t squeeze or bend.
The design can take the 3D constructions of two proteins and converts individuals structures into 3D graphs that can be processed by the neural network. Proteins are shaped from chains of amino acids, and every of individuals amino acids is represented by a node in the graph.
The researchers integrated geometric awareness into the design, so it understands how objects can adjust if they are rotated or translated in 3D place. The product also has mathematical expertise constructed in that makes certain the proteins usually attach in the similar way, no matter exactly where they exist in 3D area. This is how proteins dock in the human entire body.
Making use of this details, the equipment-mastering program identifies atoms of the two proteins that are most likely to interact and type chemical reactions, regarded as binding-pocket points. Then it uses these points to place the two proteins with each other into a advanced.
“If we can have an understanding of from the proteins which person sections are probably to be these binding pocket details, then that will seize all the information we require to spot the two proteins jointly. Assuming we can locate these two sets of factors, then we can just obtain out how to rotate and translate the proteins so a person established matches the other established,” Ganea points out.
1 of the major issues of making this product was beating the absence of teaching info. Mainly because so minor experimental 3D knowledge for proteins exist, it was especially significant to include geometric know-how into Equidock, Ganea claims. Without having individuals geometric constraints, the model could possibly choose up untrue correlations in the dataset.
Seconds vs. several hours
After the design was experienced, the scientists as opposed it to four program procedures. Equidock is ready to forecast the closing protein elaborate right after only just one to 5 seconds. All the baselines took a great deal for a longer time, from concerning 10 minutes to an hour or far more.
In top quality actions, which compute how carefully the predicted protein complex matches the actual protein intricate, Equidock was normally comparable with the baselines, but it at times underperformed them.
“We are nevertheless lagging at the rear of one of the baselines. Our method can however be enhanced, and it can even now be valuable. It could be used in a pretty large digital screening where we want to recognize how 1000’s of proteins can interact and sort complexes. Our method could be utilised to create an preliminary established of candidates really quick, and then these could be fine-tuned with some of the a lot more exact, but slower, regular solutions,” he suggests.
In addition to applying this system with conventional versions, the group would like to incorporate unique atomic interactions into Equidock so it can make more accurate predictions. For instance, sometimes atoms in proteins will connect by means of hydrophobic interactions, which require drinking water molecules.
Their method could also be utilized to the enhancement of little, drug-like molecules, Ganea says. These molecules bind with protein surfaces in distinct techniques, so fast pinpointing how that attachment happens could shorten the drug improvement timeline.
In the upcoming, they program to boost Equidock so it can make predictions for flexible protein docking. The most important hurdle there is a lack of data for education, so Ganea and his colleagues are doing the job to deliver artificial info they could use to enhance the model.
This perform was funded, in part, by the Device Discovering for Pharmaceutical Discovery and Synthesis consortium, the Swiss National Science Foundation, the Abdul Latif Jameel Clinic for Machine Studying in Well being, the DTRA Discovery of Clinical Countermeasures Towards New and Rising (DOMANE) threats system, and the DARPA Accelerated Molecular Discovery plan.





